Le guide de la médecine et de la santé
en Afrique francophone
Suivez-nous :
 Identifiez-vous | Inscription
Identifiez-vous | Inscription
Suivez-nous :
Identifiez-vous | Inscription

Adapté de l'article de Dr Romain Boulestreau - Cardiologie et maladies vasculaires - CHU de Bordeaux - 31 juillet 2023
Les recommandations ESH (Société Européenne et Internationale d'Hypertension Artérielle, Société européenne de Néphrologie) 2023 viennent renforcer et compléter les recommandations de 2018, avec quelques nouveautés :
| Catégorie | Systolique (mmHg) | Diastolique (mmHg) | Action | |
|---|---|---|---|---|
| Optimale | < 120 | et | < 80 | Surveillance ≥ /5 ans |
| Normale | 120-129 | et/ou | 80-84 | Surv. ≥ / 3 ans (6 mois si FRCV) |
| Normale haute | 130-139 | et/ou | 85-89 | Surv. annuelle, éliminer HTA masquée par AMT/MAPA, traiter si THRCV |
| Hypertension grade 1 | 140-159 | et/ou | 90-99 | Éliminer blouse blanche par AMT/MAPA, essai RHD 3-6 mois, traiter si HRCV |
| Hypertension grade 2 | 160-179 | et/ou | 100-109 | Confirmer par AMT/MAPA en qq j/sem, traitement immédiat et contrôle sous 3 mois |
| Hypertension grade 3 | ≥ 180 | et/ou | ≥ 110 | Traitement immédiat et cible atteinte à 3 mois |
| Hypertension systolique isolée | ≥ 140 | et | < 90 | Traitement selon le grade de la PAS |
| Hypertension diastolique isolée | < 140 | et | ≥ 90 | Traitement selon le grade de la PAS |
MT = automesure tensionnelle ;
HRCV/THRCV = haut/très haut risque CV.
Utiliser la plus haute valeur des mesures standardisées effectuées au cabinet.
RHD pour tous dès tension normale haute.
Découverte ≥ 65 ans: traiter si antihypertenseurs bien tolérés. Sinon attendre grade 2.
Découverte ≥ 80 ans: traiter si ≥ grade 2.
L'HTA systolique isolée est gradée selon la même échelle des valeurs systoliques.
Dépistage rapproché chez le +50 ans.

Liste des lésions organiques dûes à l'hypertension :
L'évaluation et la quantification du risque cardiovasculaire total sont importants chez les patients hypertendus afin de :
Les « guidelines » insistent sur l'importance de prendre en considération les atteintes d'organes liées à l'HTA dans l'évaluation du risque cardiovasculaire des patients. En effet, l'inclusion des atteintes d'organes liées à l'HTA aide à identifier les patients hypertendus à haut risque ou à très haut risque CV qui pourraient, sinon, être mal classés, comme présentant un niveau de risque inférieur par la table SCORE.

À défaut, des mesures répétées de la PA doivent être prises au cabinet sur plus d'une consultation, sauf lorsque l'HTA est sévère. À chaque consultation, trois mesures de PA doivent être enregistrées, espacées de 1 à 2 minutes, et des mesures supplémentaires doivent être effectuées si les deux premières lectures diffèrent de > 10 mmHg (I,C).
Les recommandations suivantes n'ont pas changé non plus :
Les objectifs sont les mêmes que l'évaluation clinique : évaluer le niveau de risque cardiovasculaire, les atteintes d'organe cible de l'HTA et les signes orientant vers une HTA secondaire.

Le bilan initial, appelé autrefois « bilan standard de l'OMS », est à réaliser chez tous les patients hypertendus au diagnostic. Il associe une hémoglobine, HBA1C, et glycémie à jeun, un bilan lipidique complet, le niveau d'acide urique, le niveau de créatininémie / le DFG, la kaliémie natrémie calcémie, un RAC (ratio albuminurie / créatinurie, qui remplace la microalbuminurie) et un ECG.
Les autres examens (notamment ETT et épreuve d'effort) ne sont recommandés que sur point d'appel lors de bilan initial (Table 6 ci-dessous).

Un des objectifs principaux de cette évaluation est de déterminer si le patient appartient à l’une des catégories suivantes, associées à une prévalence importante d’HTA secondaire :

Si le patient correspond à l'une de ces catégories, il devra bénéficier d'un bilan d'HTA secondaire complet auprès d'un praticien formé.
Vous trouverez ci-dessous 3 infographies sur la présentation typique d'un patient porteur d'un hyperaldostéronisme primaire, d'une sténose athéromateuse des artères rénales, ou d'une fibrodyslplasie des artères rénales.
Infographies : Profils caractéristiques de patients présentant une HTA secondaire
.")
FIGURE 8 A Maladie athérosclérotique rénovatrice (ARVD).
(a) La prévalence de la MRAV diffère considérablement entre les populations étudiées - dans une cohorte basée sur la population âgée de plus de 65 ans, la MRAV (définie comme une sténose de plus de 60 %) a été identifiée dans 6,8 % des cas. Chez les hypertendus, la prévalence de la MAVD est probablement de l'ordre de 1 % chez les patients souffrant d'hypertension légère, mais peut atteindre 14 à 24 % chez les patients souffrant d'hypertension sévère ou résistante.
(b) Compte tenu de l'association fréquente avec des lésions athérosclérotiques dans d'autres lits artériels, un bilan cardiovasculaire doit être envisagé.
(c) La prise en charge médicale de l'ARVD doit viser à réduire le risque CV et à protéger la fonction rénale ; le contrôle de l'hypertension est un objectif primordial. En ce qui concerne le traitement antihypertenseur, un IEC ou un ARA est considéré comme une option de première intention (contre-indiqué en cas de sténose bilatérale de l'artère rénale ou de sténose dans un rein solitaire).
(d) Des données d'observation ont montré que la pose d'une endoprothèse dans l'artère rénale, en plus du traitement médical, est associée à une amélioration de la fonction rénale et CV.
.")
FIGURE 8 B Dysplasie fibromusculaire (DFM)
(a) La DFM survient principalement chez les femmes jeunes ou d'âge moyen. Cependant, elle peut être diagnostiquée à tout âge, tant chez les femmes que chez les hommes. La DFM rénale est la deuxième cause d'hypertension rénovasculaire après la sténose athérosclérotique de l'artère rénale.
(b) Deux sous-types de fièvre aphteuse ont été décrits : la fièvre aphteuse multifocale (80-90% des cas) et la fièvre aphteuse focale (10-20% des cas). La lésion caractéristique de la fièvre aphteuse multifocale est le "chapelet de perles", caractérisé par l'alternance de zones de sténose et de dilatation dans les parties médiane et distale de l'artère. La fièvre aphteuse focale se caractérise par une sténose focale de longueur variable, qui peut se produire dans n'importe quelle partie de l'artère et nécessite l'exclusion de l'athérosclérose, des artériopathies inflammatoires ou génétiques.
(c) Dans une méta-analyse, le taux de guérison de l'hypertension après angioplastie était de 36 % (intervalle 14-85 %), mais il peut être beaucoup plus élevé chez les patients plus jeunes souffrant d'une hypertension récente. L'angioplastie doit également être envisagée chez les patients présentant une fièvre aphteuse rénale et une hypertension résistante.
(d) Des cas de plicature et de fracture de stent ont été rapportés dans le cadre de la fièvre aphteuse rénale. En conséquence, la pose d'une endoprothèse n'est généralement pas recommandée dans la DFM rénale et est réservée au traitement d'une dissection per-procédurale limitant le flux ou en cas d'anévrisme de l'artère rénale.
(e) Dans plus de 50 % des cas, les patients atteints de fièvre aphteuse rénale présentent des lésions dans un ou plusieurs autres lits artériels (fièvre aphteuse multivaisseaux). Les patients atteints de fièvre aphteuse présentent également souvent des dissections artérielles, des anévrismes ou une tortuosité artérielle marquée. Pour ces raisons, il est recommandé d'effectuer au moins une fois dans la vie une angio-TDM de la tête au bassin ou, en cas de contre-indication, une angiographie par RM chez tous les patients atteints de fièvre aphteuse.
.")
FIGURE 8 C Aldostéronisme primaire (AP).
(a) Dépend de la population examinée - varie de 3,2 % à 12,7 % en pratique primaire et de 1 % à 30 % dans les centres de référence ; la prévalence augmente avec la sévérité de l'hypertension jusqu'à 20 %.
(b) La prévalence de l'AP chez les patients présentant un incidentalome surrénalien varie de 1,6 % à 4,3 %.
(c) L'ARR nécessite au moins la normalisation du potassium plasmatique et l'interruption du traitement existant à base de spironolactone et de BB.
(d) Dans l'ensemble, l'ITS assis semble fiable et moins compliqué que le FST et le SLT. Le CCT peut être une bonne alternative chez les patients présentant un risque de surcharge hydrique (patients souffrant d'insuffisance rénale ou d'insuffisance cardiaque).
(e) Bien que la majorité des cas d'AP soient sporadiques, jusqu'à 5 % des patients peuvent présenter une forme familiale de la maladie. Un test génétique doit être effectué chez tous les patients atteints d'une forme précoce d'AP (c'est-à-dire âgés de moins de 20 ans), quelle que soit la gravité du phénotype clinique, et chez les patients ayant des antécédents familiaux d'AP.
(f) Les ARM stéroïdiens sont le traitement de choix de l'AP chez les patients présentant une maladie surrénalienne bilatérale ou une maladie unilatérale qui ne peut être traitée chirurgicalement.

Des interventions sur le mode de vie sont recommandées pour tous les patients à partir d'une pression artérielle normal-haute (> 130 / 80 mmHg). Le seuil nécessitant de démarrer un traitement pharmacologique a été ramené à 140/90 mmHg pour la majorité des patients, sauf pour les patients de plus de 80 ans, où il est fixé à 160/90 mmHg.
/!\ Pour les patients en prévention secondaire, en particulier les coronariens, il est recommandé de démarrer un traitement si la pression artérielle reste > 130 et/ou 80 mmHg de systolique et diastolique malgré les hygiéno-diététiques (Recommandation IA) /!\

Pour les patients de 18 à 64 ans : moins de 130/80 mmHg (IA) en consultation
Pour les patients de 64 à 79 ans : moins de 140/80 mmHg, voire moins de 130/80 mmHg si le traitement est bien toléré.
Pour les patients de plus de 80 ans :
Ne pas viser moins de 120/70 mmHg
Des modifications du mode de vie peuvent prévenir ou retarder l'apparition de l'HTA et réduire le risque CV, ainsi que prévenir le besoin d'un traitement médicamenteux chez les patients présentant une hypertension de grade 1. Elles doivent être systématiques.
Cependant, une intervention sur le style de vie ne devrait jamais retarder le début du traitement médicamenteux chez les patients présentant une atteinte d'organe liée à l'HTA, ou présentant un risque CV élevé.
L'algorithme décisionnel a été développé pour fournir une recommandation de traitement simple et pragmatique pour le traitement de l'HTA, basé sur quelques principes et recommandations clés. Celui-ci est comparable à l'algorithme de 2018 de part son efficacité :
Nouveautés des recommandations 2023 :



Avec les nouveaux traitements anticancéreux, la survie de cette population s'améliore. Ces patients sont à haut risque cardiovasculaire, entrainant un nouveau domaine pour le cardiologue, y compris lors de la prise en charge de l'hypertension artérielle.
Les recommandations 2023 s'y consacrent en profondeur, avec plusieurs messages clés :
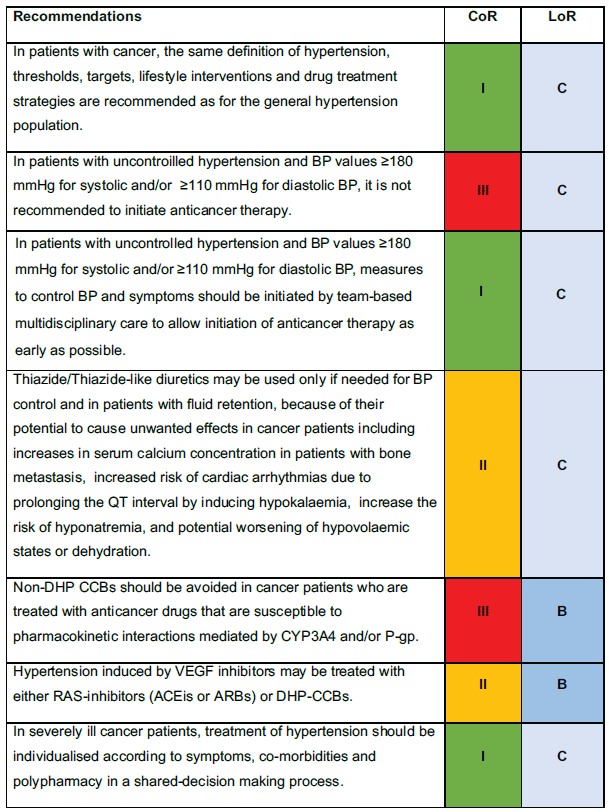
Source : Recommandations 2023 pour la prise en charge de l’HTA - Dr Romain Boulestreau - Cardio-online. Nous vous invitons à consulter le document original : Recommandations 2023 pour la prise en charge de l’hypertension artérielle
Consultez les Recommandations 2023 en texte intégral : « 2023 ESH Guidelines for the management of arterial hypertension » - Endorsed by the International Society of Hypertension (ISH) and the European Renal Association (ERA) - (PDF en anglais sur https://www.portailvasculaire.fr/)

Traitement de l'HTA essentielle chez les patients insuffisamment contrôlés par l'administration d'énalapril 20 mg seul. L'association fixe Zanextra® 20 mg/10 mg ne doit pas être utilisée en initiation de traitement de l'hypertension.
Consultez le Résumé des Caractéristiques Produit (RCP) de Zanextra® 20 mg / 10 mg

Traitement de l'HTA essentielle en substitution chez les patients dont la pression artérielle est correctement contrôlée par l'administration séparée d'énalapril 20 mg et de lercanidipine 20 mg pris simultanément.
Consultez le Résumé des Caractéristiques Produit (RCP) de Zanextra® 20 mg / 20 mg


Zanextra® 20 mg/10mg, comprimé pelliculé
Chaque comprimé contient 20 mg de maléate d’énalapril (équivalent à 15,29 mg d'énalapril) et 10 mg de chlorhydrate de lercanidipine (équivalent à 9,44 mg de lercanidipine).
Excipient à effet notoire : Chaque comprimé contient 92,0 mg de lactose monohydraté.
Pour la liste complète des excipients, voir rubrique 6.1.
Comprimé pelliculé.
Comprimé de 8.5 mm, jaune, rond et biconvexe.
Traitement de l’hypertension artérielle essentielle chez les patients insuffisamment contrôlés par l’administration de l’énalapril 20 mg seul.
L’association fixe ZANEXTRA® 20 mg/10 mg ne doit pas être utilisée en initiation de traitement de l’hypertension.
Chez les patients dont la pression artérielle est insuffisamment contrôlée par l’énalapril 20 mg seul, la posologie d’énalapril peut être augmentée, en monothérapie ou le traitement peut être remplacé par ZANEXTRA® 20 mg/10 mg.
L’adaptation individuelle de la posologie de chacun des composants est recommandée. Le passage direct de la monothérapie à l’association fixe peut être envisagé s’il est cliniquement justifié.
La posologie usuelle recommandée est d’un comprimé par jour à prendre au moins 15 minutes avant le repas.
La posologie doit être adaptée à la fonction rénale du patient (voir « Utilisation en cas d’insuffisance rénale »).
ZANEXTRA® est contre-indiqué chez les patients atteints d’une insuffisance rénale sévère (clairance de la créatinine < 30 ml/min) ou chez les patients sous hémodialyse (voir rubriques 4.3 et 4.4). Le traitement doit être initié avec une prudence particulière chez les patients atteints d’insuffisance rénale légère à modérée.
ZANEXTRA® est contre-indiqué en cas d’insuffisance hépatique sévère. Le traitement doit être initié avec prudence chez les patients atteints d’insuffisance hépatique légère à modérée.
Dans cette indication, l’utilisation de ZANEXTRA® dans la population pédiatrique n’est pas recommandée.
Précautions à prendre avant l’administration de ce médicament :
L’association de Zanextra® avec des médicaments contenant de l’aliskiren est contre-indiquée chez les patients diabétiques ou insuffisants rénaux (DFG < 60 ml/min/1,73 m2) (voir rubriques 4.5 et 5.1).
Une hypotension symptomatique est rarement observée en cas d’hypertension non-compliquée. Une hypotension symptomatique est plus susceptible de survenir chez les patients hypertendus traités par énalapril en cas de déplétion hydrique préalable (traitement par diurétique, régime hyposodé, dialyse, diarrhées ou vomissements) (voir rubrique 4.5). Chez les patients présentant une insuffisance cardiaque, avec ou sans insuffisance rénale associée, des cas d’hypotension symptomatique ont été observés. Le risque est plus important chez les patients présentant des degrés plus sévères d’insuffisance cardiaque, se caractérisant par l’utilisation de doses élevées de diurétique de l’anse, une hyponatrémie ou une insuffisance rénale fonctionnelle.
Chez ces patients, le traitement doit être commencé sous contrôle médical et les patients doivent être suivis étroitement à chaque fois que la posologie d’énalapril et/ou du diurétique est ajustée. Des précautions similaires doivent s’appliquer aux patients présentant une cardiopathie ischémique ou une maladie vasculaire cérébrale, chez lesquels une diminution excessive de la pression artérielle pourrait entraîner un infarctus du myocarde ou un accident vasculaire cérébral.
Si une hypotension survient, le patient doit être allongé et, si nécessaire, recevoir une perfusion intraveineuse de solution saline isotonique. Une réaction hypotensive transitoire ne constitue pas une contre-indication à la poursuite du traitement, qui peut être continué sans difficulté, dès l’augmentation de la pression artérielle, après remplissage vasculaire.
Chez certains patients atteints d’insuffisance cardiaque mais présentant une pression artérielle normale ou faible, une diminution supplémentaire de la pression artérielle peut survenir avec l’énalapril. Cet effet est prévisible et ne constitue généralement pas une raison pour interrompre le traitement. Si l’hypotension devient symptomatique, une réduction de la posologie et/ou l’interruption du diurétique et/ou de l’énalapril peuvent être nécessaires.
La lercanidipine doit être administrée avec prudence chez les patients présentant une dysfonction du nœud sinusal (lorsqu’un stimulateur cardiaque n’a pas été installé).
Même si des études hémodynamiques contrôlées n’ont révélé aucune altération de la fonction ventriculaire, des précautions doivent être prises chez des patients ayant une dysfonction ventriculaire gauche.
Les données suggèrent que les patients atteints de cardiopathie ischémique présentent un risque cardiovasculaire élevé sous traitement par certaines dihydropyridines à courte durée d’action. Bien que la lercanidipine possède une longue durée d’action, la prudence est recommandée chez ces patients. Dans de rares cas, certaines dihydropyridines peuvent entraîner des douleurs précordiales ou un angor. Dans de très rares cas, des patients ayant un angor préexistant peuvent présenter une augmentation de la fréquence, de la durée ou de la gravité des crises angineuses. Des cas isolés d’infarctus du myocarde peuvent être observés (voir rubrique 4.8).
Le traitement avec l’énalapril doit être initié avec prudence particulière chez les patients atteints d’insuffisance rénale légère à modérée. Le contrôle systématique de la kaliémie et de la créatinémie au cours d’un traitement par énalapril, fait partie de la surveillance normale chez ces patients.
Des cas d’insuffisance rénale ont été rapportés avec l’énalapril, surtout chez des patients ayant une insuffisance cardiaque sévère ou une maladie rénale sous-jacente, y compris une sténose de l’artère rénale. Si elle est diagnostiquée rapidement et traitée de façon appropriée, l’insuffisance rénale, sous énalapril, est habituellement réversible.
Certains patients hypertendus, sans altération rénale préexistante, ont présenté des augmentations de l’urémie et de la créatininémie au cours d’utilisation concomitante d’énalapril et d’un diurétique. Une diminution de la dose d’énalapril et/ou l’arrêt du diurétique peuvent être nécessaires. Cette situation devrait évoquer la possibilité d’une sténose sous-jacente des artères rénales (voir rubrique 4.4 Hypertension réno-vasculaire).
Il existe un risque accru d’hypotension et d’insuffisance rénale, si les patients, présentant une sténose bilatérale de l’artère rénale ou une sténose de l'artère de leur seul rein fonctionnel sont traités avec des IEC. La perte de la fonction rénale peut survenir avec des modifications même mineures de la créatinémie.
Chez ces patients, le traitement doit être initié sous surveillance médicale étroite, avec de faibles doses puis une adaptation prudente de la posologie et un suivi de la fonction rénale.
Il n’y a aucune expérience concernant l’utilisation de lercanidipine ou d’énalapril chez les patients ayant récemment subi une transplantation rénale. Par conséquent, le traitement de ces patients par Zanextra® est déconseillé.
L’effet antihypertenseur de la lercanidipine peut être potentialisé chez les patients ayant une dysfonction hépatique.
Rarement, un syndrome débutant par un ictère choléstatique ou une hépatite et évoluant vers une nécrose hépatique fulminante (parfois fatale) a été observé avec un traitement par les IEC. Le mécanisme de ce syndrome n’a pas été clairement élucidé. Les patients qui développent un ictère ou une augmentation marquée des enzymes hépatiques avec les IEC, doivent arrêter la prise de l’IEC et recevoir un suivi médical approprié.
Chez les patients sous dialyse péritonéale, la lercanidipine a été associée à des liquides de dialyse troubles. La turbidité est due à une augmentation de la concentration des triglycérides dans le liquide de dialyse. Bien que le mécanisme ne soit pas expliqué, la turbidité s’améliore après l’arrêt de la lercanidipine. Il s’agit d’un effet important à reconnaître car un liquide de dialyse trouble peut être confondu avec une péritonite infectieuse et causer inutilement une hospitalisation et une administration d’antibiotiques.
Des cas de neutropénie, d’agranulocytose, de thrombocytopénie ou d’anémie ont été rapportés chez des patients recevant des IEC. La neutropénie survient rarement chez les patients ayant une fonction rénale normale et ne présentant aucun facteur de risque particulier. L’énalapril doit être administré avec une extrême prudence chez les patients ayant une collagénose vasculaire, recevant un traitement immunosuppresseur, de l’allopurinol, de la procaïnamide, une association ou présentant de ces facteurs aggravants particulièrement en cas d’altération préexistante de la fonction rénale. Certains de ces patients ont développé des infections graves qui, dans quelques cas, n’ont pas répondu à un traitement antibiotique intensif. Si l’énalapril est utilisé chez ces patients, un contrôle régulier de la numération leucocytaire est recommandé et les patients doivent signaler tout signe d’infection à leur médecin.
Un œdème angioneurotique avec atteinte du visage, des extrémités, des lèvres, de la langue, de la glotte et/ou du larynx a été rapporté chez des patients traités par IEC, dont l’énalapril. Ceci peut survenir à n’importe quel moment au cours du traitement. Dans de tels cas, l’énalapril doit être arrêté immédiatement et une surveillance appropriée doit être mise en place afin de s’assurer de la disparition complète des symptômes avant de le laisser partir le patient.
Même dans les cas où seul un gonflement de la langue, sans détresse respiratoire, est observé, une observation prolongée peut être nécessaire pour ces patients car un traitement, par antihistaminiques ou corticostéroïdes, peut ne pas être suffisant.
Dans de très rares cas, des décès ont été rapportés à la suite d'angiœdème avec atteinte laryngée ou linguale. Les patients présentant un œdème de la langue, de la glotte ou du larynx sont susceptibles de présenter une obstruction des voies respiratoires, en particulier ceux ayant des antécédents de chirurgie des voies respiratoires.
En cas d’atteinte de la langue, de la glotte ou du larynx, susceptible d’entraîner une obstruction des voies aériennes, un traitement approprié qui peut comporter une injection sous-cutanée d’adrénaline à 1:1000 (0,3 ml à 0,5 ml) et/ou des mesures visant à assurer la liberté des voies aériennes doivent être entrepris rapidement.
Une incidence plus élevée d’angiœdème sous traitement par IEC a été rapportée chez les populations noires.
Les patients ayant un antécédent d’angiœdème non-lié à la prise d’IEC, peuvent avoir un risque accru d’angiœdème s’ils reçoivent un IEC (voir rubrique 4.3).
L’association des inhibiteurs de l’enzyme de conversion avec le sacubitril/valsartan est contre-indiquée en raison d’un risque accru d’angiœdème. Un traitement par sacubitril/valsartan ne doit être initié que 36 heures après la prise de la dernière dose d’énalapril. Un traitement par énalapril ne doit être instauré que 36 heures après la dernière dose de sacubitril/valsartan.
L’association des inhibiteurs de l’enzyme de conversion avec le racécadotril, les inhibiteurs mTor (ex sirolimus, évérolimus, temsirolimus) et la vildagliptine peuvent provoquer une augmentation du risque d’angiœdème (gonflement des voies respiratoires ou de la langue avec ou sans difficulté respiratoire) (voir rubrique 4.5). Des précautions doivent être prises lorsque ces médicaments doivent être initiés chez un patient qui prend un inhibiteur de l’enzyme de conversion.
Dans de rares cas, des réactions anaphylactoïdes avec menace du pronostic vital se sont produites au cours d’un traitement de désensibilisation aux venins d’hyménoptères chez des patients prenant concomitamment un IEC. Ces réactions peuvent être évitées en arrêtant temporairement le traitement par l’IEC avant chaque désensibilisation.
Dans de rares cas, des réactions anaphylactoïdes avec menace du pronostic vital se sont produites lors d’aphérèses des lipoprotéines de basse densité (LDL) avec du sulfate de dextran chez des patients prenant concomitamment un IEC. Ces réactions peuvent être évitées en arrêtant temporairement le traitement par l’IEC avant chaque aphérèse.
Les patients diabétiques traités par antidiabétiques oraux ou par insuline, débutant un traitement par IEC, doivent être informés de veiller particulièrement au risque d’hypoglycémie, spécialement au cours du premier mois de traitement par l’association de ces deux médicaments (voir rubrique 4.5).
Une toux a été rapportée avec l’utilisation d’IEC. Typiquement, cette toux est non productive, persistante et disparaît à l’arrêt du traitement. Une toux induite par un IEC doit être également envisagée lors du diagnostic différentiel d’une toux.
Chez les patients ayant une intervention chirurgicale majeure ou au cours d'une anesthésie pratiquée avec des agents hypotenseurs, l'énalapril bloque la formation d'angiotensine II secondaire à la sécrétion compensatrice de rénine. Si une hypotension survient et est considérée comme due à ce mécanisme, elle peut être corrigée par le remplissage vasculaire.
Les IEC peuvent provoquer une augmentation de la kaliémie car ils inhibent la libération d’aldostérone. L’effet n’est en général pas significatif chez les patients avec une fonction rénale normale. Cependant, chez les patients ayant une insuffisance rénale et/ou qui prennent des suppléments potassiques (incluant des substituts de sel contenant du potassium), des diurétiques épargneurs de potassium, du triméthoprime ou du cotrimoxazole (triméthoprime/sulfméthoxazole) ou plus particulièrement des antagonistes de l’aldostérone ou des antagonistes des récepteurs de l’angiotensine, des hyperkaliémies peuvent arriver.
Les diurétiques épargneurs de potassium et les antagonistes des récepteurs de l’angiotensine doivent être utilisés avec précaution chez les patients qui ont un traitement par IEC ; la kaliémie et la fonction rénale doivent être contrôlées chez ces patients (voir rubrique 4.5).
L’association du lithium et de l’énalapril n’est généralement pas recommandée (voir rubrique 4.3).
Il est établi que l’association d’inhibiteurs de l’enzyme de conversion (IEC), d’antagonistes des récepteurs de l’angiotensine II (ARA II) ou d’aliskiren augmente le risque d’hypotension, d’hyperkaliémie et d’altération de la fonction rénale (incluant le risque d’insuffisance rénale aiguë). En conséquence, le double blocage du SRAA par l’association d’IEC, d’ARA II ou d’aliskiren n’est pas recommandé (voir rubriques 4.5 et 5.1).
Néanmoins, si une telle association est considérée comme absolument nécessaire, elle ne pourra se faire que sous la surveillance d’un spécialiste et avec un contrôle étroit et fréquent de la fonction rénale, de l’ionogramme sanguin et de la pression artérielle.
L’association d’un IEC avec un ARA II est contre-indiquée chez les patients avec une néphropathie diabétique.
Les inducteurs du cytochrome CYP3A4 tels que les anticonvulsivants (par exemple, la phénytoïne, la carbamazépine) et la rifampicine peuvent réduire les concentrations plasmatiques de lercanidipine. Par conséquent, l'efficacité de la lercanidipine peut être inférieure à celle attendue (voir rubrique 4.5).
Comme avec les autres IEC, l’énalapril semble être moins efficace pour diminuer la pression artérielle chez les patients noirs que chez les autres patients. Ceci peut être éventuellement expliqué par des concentrations plasmatiques de rénine souvent plus faibles dans la population noire hypertendue.
Zanextra® est contre-indiqué pendant la grossesse.
Les IEC, tels que l’énalapril ne doivent pas être débutés au cours de la grossesse. A moins que le traitement par IEC ne soit considéré comme essentiel, il est recommandé aux patientes qui envisagent une grossesse de modifier leur traitement antihypertenseur pour un médicament ayant un profil de sécurité bien établi pendant la grossesse. En cas de diagnostic de grossesse, le traitement par IEC doit être arrêté immédiatement et si nécessaire, un traitement alternatif sera débuté (voir rubriques 4.3 et 4.6).
Le traitement par lercanidipine est aussi déconseillé pendant la grossesse et chez les femmes en âge de procréer (voir rubrique 4.6).
Zanextra® est déconseillé pendant l’allaitement (voir rubrique 4.6).
En l’absence de données cliniques, l’efficacité et la sécurité d’emploi n’ont pas été démontrées chez les enfants.
L’alcool doit être évité car il peut potentialiser l’effet des antihypertenseurs vasodilatateurs (voir rubrique 4.5).
En raison de la présence de lactose, ce médicament est contre-indiqué en cas de galactosémie congénitale, de syndrome de malabsorption du glucose et du galactose ou de déficit en lactase.
L’effet antihypertenseur de Zanextra pourrait être potentialisé par d’autres anti-hypertenseurs tels que les diurétiques, les bêta-bloquants, les alpha-bloquants et autres substances.
De plus, les interactions suivantes ont été observées avec l’un ou l’autre des constituants de cette association fixe.
Médicaments augmentant le risque d’angiœdème
L’association des IEC avec le sacubitril/valsartan est contre-indiquée en raison d’un risque plus élevé d’angiœdème (voir rubriques 4.3 et 4.4).
L’association des IEC avec le racécadotril, les inhibiteurs mTOR (ex sirolimus, évérolimus, temsirolimus) et la vildagliptine peuvent provoquer une augmentation du risque d’angiœdème (voir rubrique 4.4).
Les données issues des essais cliniques ont montré que le double blocage du système rénine-angiotensine-aldostérone (SRAA) par l’utilisation concomitante d’inhibiteur de l’enzyme de conversion, d’antagonistes des récepteurs de l’angiotensine II ou d’aliskiren est associé à une fréquence plus élevée d’évènements indésirables, tels que l’hypotension, l’hyperkaliémie et l’altération de la fonction rénale (incluant l’insuffisance rénale aiguë) en comparaison à l’utilisation d’un seul médicament agissant sur le SRAA (voir rubriques 4.3, 4.4 et 5.1).
Bien que la kaliémie reste habituellement dans les limites normales, une hyperkaliémie peut survenir chez certains patients traités par énalapril. Les diurétiques épargneurs potassiques (ex. spironolactone, triamtérène ou amiloride), les suppléments de potassium ou les sels de remplacement contenant du potassium peuvent entraîner des augmentations significatives de la kaliémie.
Il convient également de faire preuve de prudence lors de l’administration d’énalapril avec d’autres médicaments hyperkaliémiants, tels que le triméthoprime et le cotrimoxazole (triméthoprime/sulfaméthoxazole) car le triméthoprime agit comme un diurétique épargneur de potassium tel que l’amiloride. Par conséquent, l’association de l’énalapril avec les médicaments susmentionnés n’est pas recommandée. Si une utilisation concomitante est indiquée, elle doit se faire avec précaution et être accompagnée d’une surveillance fréquente de la kaliémie.
L’association des IEC avec la ciclosporine peut provoquer une hyperkaliémie.
La surveillance de la kaliémie est recommandée.
L’association des IEC avec l’héparine peut provoquer une hyperkaliémie.
La surveillance de la kaliémie est recommandée.
Un traitement préalable avec des doses élevées de diurétiques peut provoquer une hypovolémie et un risque d’hypotension lors de l’instauration d’un traitement par énalapril (voir rubrique 4.4). Les effets hypotenseurs peuvent être diminués par un arrêt du diurétique, en augmentant la volémie, par l’apport en sel ou en instaurant le traitement avec une dose faible d’énalapril.
L’administration concomitante d’énalapril et d’autres antihypertenseurs peut augmenter les effets hypotenseurs de l’énalapril. L’administration concomitante de trinitrine et d’autres nitrates ou d’autres vasodilatateurs peut réduire davantage la pression artérielle.
Les effets réversibles suivants ont été rapportés lors de l’administration simultanée de lithium et d’IEC : augmentation des concentrations sériques de lithium et effets toxiques. L’administration concomitante de diurétiques thiazidiques peut augmenter davantage le taux de lithium et accroître le risque de toxicité avec les IEC. L’administration d’énalapril et de lithium est déconseillée. Cependant, si cette association est jugée nécessaire, une surveillance étroite des concentrations sériques de lithium doit être mise en place (voir rubrique 4.4).
L’administration concomitante de certains médicaments anesthésiques, d’antidépresseurs tricycliques, de neuroleptiques et d’IEC peut entraîner une réduction supplémentaire de la pression artérielle (voir rubrique 4.4).
Les anti-inflammatoires non stéroïdiens (AINS) incluant les inhibiteurs sélectifs de la cyclooxygénase-2 (inhibiteurs de la COX-2) peuvent réduire l’effet des diurétiques et autres médicaments antihypertenseurs. Par conséquent, les effets antihypertenseurs des ARA II ou des IEC peuvent être diminués par les AINS notamment les inhibiteurs sélectifs de la COX-2.
La co-administration d’AINS (incluant les inhibiteurs de la COX-2) et d’ARA II ou d’IEC exerce un effet additif sur l’augmentation de la kaliémie, ce qui peut entraîner une détérioration de la fonction rénale. Cet effet est généralement réversible. Rarement, une insuffisance rénale aiguë peut survenir, particulièrement chez les patients ayant une fonction rénale altérée tels que les patients âgés ou présentant une hypoglycémie (y compris ceux sous traitement diurétique). Par conséquent, l’association doit être administrée avec prudence chez les patients ayant une fonction rénale altérée. Les patients doivent être hydratés en conséquence et leur fonction rénale surveillée, après l’initiation d’un traitement concomitant, et de façon régulière ensuite.
Des réactions nitritoïdes (symptômes incluant bouffées vasomotrices du visage, nausées, vomissements et hypotension) ont été notifiées dans de rares cas chez des patients recevant un traitement par des sels d’or injectables (aurothiomalate sodique) et un traitement concomitant par un IEC, dont l’énalapril.
Les sympathomimétiques peuvent réduire les effets antihypertenseurs des IEC.
Des études épidémiologiques ont suggéré que l’administration concomitante d’IEC et d’antidiabétiques (insuline, antidiabétiques oraux) peut entraîner un effet hypoglycémiant plus important, avec un risque d’hypoglycémie. Ces cas sont plus susceptibles de survenir dans les premières semaines de la co-administration, ainsi que chez des patients ayant une insuffisance rénale (voir rubriques 4.4 et 4.8).
L’alcool renforce l’effet hypotenseur des IEC.
L’énalapril peut être administré sans risque avec de l’acide acétylsalicylique (à doses cardiologiques), des thrombolytiques, et des β-bloquants.
Associations contre-indiquées
La lercanidipine est métabolisée par l’enzyme CYP3A4 et, par conséquent, les inhibiteurs du cytochrome CYP3A4 administrés simultanément peuvent interagir avec le métabolisme et l’élimination de la lercanidipine. Une étude d'interaction avec le kétoconazole, un puissant inhibiteur du cytochrome CYP3A4, a montré une augmentation considérable des concentrations plasmatiques de lercanidipine (multiplication par 15 de l'ASC et par 8 de la Cmax pour l'énantiomère S-lercanidipine).
L’administration de lercanidipine avec des inhibiteurs du CYP3A4 (par exemple kétoconazole, itraconazole, ritonavir, érythromycine, troléandomycine, clarithromycine) doit être évitée (voir rubrique 4.3).
Une élévation des concentrations plasmatiques de lercanidipine et de ciclosporine a été observée après administration concomitante des 2 principes actifs. Une étude chez de jeunes volontaires sains a montré que, lorsque la ciclosporine était administrée 3 heures après la prise de lercanidipine, les concentrations plasmatiques de lercanidipine n’avaient pas changé, tandis que l’ASC de la ciclosporine augmentait de 27%. Cependant, l'administration concomitante de lercanidipine et de ciclosporine a provoqué une augmentation de 3 fois les concentrations plasmatiques de lercanidipine et une augmentation de 21% de l'ASC de la ciclosporine.
La ciclosporine et la lercanidipine ne doivent pas être administrées ensemble (voir rubrique 4.3).
Comme pour les autres dihydropyridines, le métabolisme de la lercanidipine est sensible à l’inhibition du métabolisme causé par le pamplemousse ou du jus de pamplemousse, ce qui entraîne une augmentation de la biodisponibilité systémique et de l’effet hypotenseur de la lercanidipine.
La lercanidipine ne doit pas être prise avec du pamplemousse ou du jus de pamplemousse (voir rubrique 4.3).
En cas d’administration concomitante de lercanidipine et d’inducteurs du CYP3A4, comme les anticonvulsivants (ex : phénytoïne, phénobarbital, carbamazépine) et la rifampicine, une attention particulière est requise car l’effet anti-hypertenseur de la lercanidipine peut être diminué. La pression artérielle doit donc être contrôlée plus fréquemment que d’habitude (voir rubrique 4.4).
L’alcool doit être évité car qu’il peut potentialiser l’effet des antihypertenseurs vasodilatateurs (voir rubrique 4.4).
Une prudence est nécessaire en cas d’association de lercanidipine avec des substrats du CYP3A4 comme la terfénadine, l’astémizole, les antiarythmiques de classe III (ex : amiodarone, quinidine, sotalol).
Lorsque la lercanidipine, à la dose de 20 mg, a été administrée en même temps que du midazolam par voie orale, chez des volontaires âgés, l'absorption de la lercanidipine a été augmentée (d'environ 40%) et le taux d'absorption a été diminué (le Tmax a été retardé de 1,75 à 3 heures). Les concentrations de midazolam n'ont pas été modifiées.
Lors de l’administration simultanée de lercanidipine et de métoprolol, un b-bloquant éliminé principalement par voie hépatique, la biodisponibilité du métoprolol est restée inchangée, alors que celle de la lercanidipine a été réduite de 50%. Cet effet peut être causé par la diminution du débit sanguin hépatique par les β-bloquants et pourrait donc également se produire avec d’autres médicaments de cette classe thérapeutique. Par conséquent, l’administration concomitante de lercanidipine et des β-bloquants est possible mais une adaptation posologique peut être nécessaire.
L’administration simultanée de 20 mg de lercanidipine chez des patients sous traitement chronique par la β-méthyldigoxine n’a mis en évidence aucun signe d’interaction pharmacocinétique. Cependant, une augmentation moyenne de 33% de la Cmax de la digoxine a été observée, alors que ni l’ASC ni la clairance rénale n’ont été significativement modifiées. Les signes cliniques d’une intoxication à la digoxine doivent être étroitement surveillés chez les patients sous traitement concomitant par digoxine.
Une étude d’interaction avec la fluoxétine (un inhibiteur du CYP2D6 et du CYP3A4), réalisée chez des volontaires sains âgés de 65 ± 7 ans (moyenne, écart-type), n’a pas montré de modification clinique significative de la pharmacocinétique de la lercanidipine.
L’administration concomitante d’une dose quotidienne de 800 mg de cimétidine n’entraîne pas de modification significative des concentrations plasmatiques de lercanidipine. Cependant, à doses plus élevées, une prudence est requise car la biodisponibilité de la lercanidipine, et par conséquent son effet hypotenseur, peuvent être augmentés.
L’administration répétée d’une dose de 20 mg de lercanidipine simultanément à 40 mg de simvastatine, n’a pas entraîné de modification significative de l’ASC de la lercanidipine mais a entraîné une augmentation de 56% de l’ASC de la simvastatine et une augmentation de 28% de l’ASC de son métabolite actif, le β-hydroxyacide. Il est peu probable que de telles modifications soient pertinentes sur le plan clinique. Aucune interaction n’est attendue si la lercanidipine est administrée le matin et la simvastatine le soir, tel qu’il est indiqué pour ce dernier.
L’administration simultanée de 20 mg de lercanidipine à des volontaires sains à jeun n’a pas modifié la pharmacocinétique de la warfarine.
La lercanidipine a été administrée en toute sécurité avec des diurétiques et des inhibiteurs de l’enzyme de conversion.
Comme avec tous les médicaments antihypertenseurs, une augmentation des effets hypotenseurs peut être observés quand la lercanidipine est administrée avec d’autres médicaments agissant sur la pression artérielle tels que les alpha-bloquants utilisés dans le traitement des symptômes urinaires, les antidépresseurs tricycliques et les neuroleptiques.
Au contraire, une baisse de la pression artérielle peut être observée avec des corticostéroïdes.
Les études d’interaction ont été réalisées uniquement chez l’adulte.
L’utilisation des IEC (énalapril) est déconseillée pendant le 1er trimestre de la grossesse (voir rubrique 4.4). L’utilisation des IEC (énalapril) est contre-indiquée aux 2ème et 3ème trimestres de la grossesse (voir rubriques 4.3 et 4.4).
Les données épidémiologiques disponibles concernant le risque de malformation après exposition aux IEC au 1er trimestre de la grossesse ne permettent pas de conclure. Cependant une petite augmentation du risque de malformations congénitales ne peut être exclue. A moins que le traitement par IEC ne soit considéré comme essentiel, il est recommandé aux patientes qui envisagent une grossesse de modifier leur traitement antihypertenseur pour un médicament ayant un profil de sécurité bien établi pendant la grossesse. En cas de diagnostic de grossesse, le traitement par IEC doit être immédiatement arrêté et si nécessaire, un traitement alternatif sera débuté.
L’exposition aux IEC au cours des 2ème et 3ème trimestres de la grossesse est connue pour entraîner une foetotoxicité (diminution de la fonction rénale, oligohydramnios, retard d’ossification des os du crâne) et une toxicité chez le nouveau-né (insuffisance rénale, hypotension, hyperkaliémie) (voir rubrique 5.3). Un oligohydramnios, signe probable d’une fonction rénale foetale diminuée, a été signalé et peut entraîner des contractures des membres, des déformations crânio-faciales et une hypoplasie pulmonaire. En cas d’exposition à un IEC à partir du 2ème trimestre de la grossesse, il est recommandé d’effectuer une échographie fœtale afin de vérifier la fonction rénale et les os du crâne. Les nouveau-nés de mères traitées par IEC doivent être surveillés pour déceler une éventuelle hypotension (voir rubriques 4.3 et 4.4).
Il n’y a pas de donnée sur l'utilisation de la lercanidipine pendant la grossesse. Les études réalisées chez l’animal n’ont pas mis en évidence d’effets tératogènes, mais ceux-ci ont été observés avec d’autres dihydropyridines.
L’utilisation de la lercanidipine n’est pas recommandée au cours de la grossesse ou chez les femmes en âge de procréer, en l'absence de contraception efficace (voir rubrique 4.4).
Il n’y a pas ou peu de données sur l’utilisation du maléate d’énalapril/ chlorhydrate de lercanidipine chez la femme enceinte. Les études de toxicité effectuées chez l’animal sur la reproduction sont insuffisantes (voir rubrique 5.3.).
Par conséquent, l’utilisation de Zanextra® est contre-indiquée durant les 2ème et 3ème trimestres de la grossesse. Elle est déconseillée au cours du 1er trimestre de la grossesse et chez les femmes en âge de procréer, en l'absence d'une méthode contraceptive efficace.
Des données pharmacocinétiques limitées montrent de très faibles concentrations dans le lait (voir rubrique 5.2). Bien que ces concentrations ne semblent pas avoir de réelles conséquences cliniques, l’administration d’énalapril est déconseillée chez les femmes qui allaitent des enfants prématurés et au cours des premières semaines qui suivent l’accouchement, en raison du risque hypothétique d’effets secondaires au niveau cardiovasculaire et rénal chez l’enfant et de l’expérience clinique insuffisante.
Dans le cas de l’allaitement d’un enfant plus âgé, l’administration d’énalapril peut être envisagée chez une femme qui allaite, si ce traitement est nécessaire pour la mère et que l’enfant est surveillé dans le but de détecter d’éventuels effets secondaires.
L’excrétion de la lercanidipine ou de ses métabolites dans le lait maternel est inconnue. Un risque chez le nouveau-né ou le nourrisson ne peut être exclu. La lercanidipine ne doit pas être utilisée pendant l’allaitement.
En conséquence, l’utilisation de ZANEXTRA® est déconseillée pendant l’allaitement.
Il n’y a pas de donnée disponible pour la lercanidipine. Certains inhibiteurs calciques ont été associés à des modifications biochimiques réversibles dans la région céphalique des spermatozoïdes, ce qui peut affecter la fécondation. En cas d’échecs répétés de fécondations in vitro, et en l’absence d’une autre explication, la possibilité que les inhibiteurs calciques soient à l’origine de ces échecs doit être envisagée.
L’influence de ZANEXTRA® sur l’aptitude à conduire des véhicules et à utiliser des machines est faible. Cependant, la prudence est recommandée en raison de la survenue possible de sensations vertigineuses, d’asthénie, de fatigue et rarement de somnolence (voir rubrique 4.8).
La tolérance de Zanextra® avait été évaluée dans cinq études cliniques contrôlées, en double aveugle et dans deux phases ouvertes d'extension à long terme. Au total, 1 141 patients ont reçu Zanextra® à la dose de 10 mg/10 mg, 20 mg/10 mg et 20 mg/20 mg. Les effets indésirables de cette association fixe sont comparables à ceux observés lors de l’administration de l’un ou de l’autre de ses constituants. Les effets indésirables les plus fréquemment rapportés au cours du traitement avec Zanextra® sont la toux (4,03%), les sensations vertigineuses (1,67%) et les maux de tête (1,67%).
Les effets indésirables rapportés dans les études cliniques avec Zanextra 10 mg/10 mg, 20 mg/10 mg et 20 mg/20 mg et pour lesquels il existe un lien de causalité, sont décrits dans le tableau suivant, selon le système de classe organes MedDRA et par fréquence : très fréquent (> 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), non-connus (impossibles à estimer d'après les données disponibles) :
| Affections hématologiques et du système lymphatique | |
|---|---|
| Peu fréquent | Thrombocytopénie |
| Rare | Diminution de l’hémoglobine |
| Affections du système immunitaire | |
| Rare | Hypersensibilité |
| Troubles du métabolisme et de la nutrition | |
| Peu fréquent | Hyperkaliémie |
| Affections psychiatriques | |
| Peu fréquent | Anxiété |
| Affections du système nerveux | |
| Fréquent | Sensations vertigineuses, céphalées |
| Peu fréquent | Sensations vertigineuses posturales |
| Affections de l'oreille et du labyrinthe | |
| Peu fréquent | Vertiges |
| Rare | Acouphènes |
| Affections cardiaques | |
| Peu fréquent | Tachycardie, palpitations |
| Affections vasculaires | |
| Peu fréquent | Bouffées vasomotrices, hypotension |
| Rare | Défaillance circulatoire |
| Affections respiratoires, thoraciques et médiastinales | |
| Fréquent | Toux |
| Rare | Sécheresse de la gorge, douleur oro-pharyngée |
| Affections gastro-intestinales | |
| Peu fréquent | Douleur abdominale, constipation, nausées |
| Rare | Dyspepsie, œdème des lèvres, atteinte linguale, diarrhée, sécheresse buccale, gingivite |
| Affections hépato-biliaires | |
| Peu fréquent | Augmentation ALAT et ASAT |
| Affections de la peau et du tissu sous-cutané | |
| Peu fréquent | Érythème |
| Rare | Angiœdème, gonflement du visage, dermatite, éruption cutanée, urticaire |
| Affections musculo-squelettiques et systémiques | |
| Peu fréquent | Arthralgie |
| Affections du rein et des voies urinaires | |
| Peu fréquent | Pollakiurie |
| Rare | Nycturie, polyurie |
| Affections du système de reproduction et du sein | |
| Rare | Dysfonction érectile |
| Troubles généraux et anomalies au site d'administration | |
| Peu fréquent | Asthénie, fatigue, sensation de chaleur, œdèmes périphériques |
Les effets indésirables survenus chez un seul patient sont déclarés à des fréquences rares.
Les effets indésirables observés avec l'énalapril ou la lercanidipine peuvent être aussi rapportés avec ZANEXTRA®, même s’ils n’ont pas été observés dans les essais cliniques ou pendant la période de commercialisation.
Les effets indésirables rapportés avec l’énalapril sont :
* L’incidence est comparable aux groupes placebo et aux groupes comparateurs actifs dans les essais cliniques.
Un cortège de symptômes, pouvant inclure certains ou l’ensemble des symptômes suivants a été rapporté : fièvre, sérite, vascularite, myalgie/myosite, arthralgie/arthrite, anticorps antinucléaires (ANA) positifs, élévation de la vitesse de sédimentation érythrocytaire (VSE), éosinophilie et leucocytose. Une éruption cutanée, une photosensibilité et d’autres manifestations dermatologiques peuvent survenir.
Les effets indésirables les plus fréquemment rapportés au cours d’essais cliniques et au cours de la commercialisation ont été œdèmes périphériques, céphalées, bouffées vasomotrices, tachycardie et palpitations.
1 Effets indésirables issus de notifications spontanées après commercialisation internationale.
Certaines dihydropyridines peuvent, dans de rares cas, provoquer des douleurs précordiales ou un angor. Dans de très rares cas, des patients ayant un angor préexistant peuvent présenter une augmentation de la fréquence, de la durée ou de la sévérité des crises angineuses. Des cas isolés d’infarctus du myocarde peuvent être observés.
La lercanidipine ne semble pas modifier la glycémie ou les taux lipidiques.
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration.
Depuis la commercialisation, quelques cas de surdosage intentionnel ayant nécessité une hospitalisation ont été notifiés sous énalapril/lercanidipine, avec les doses comprises entre 100 mg à 1000 mg pour chacun de ces principes actifs en une prise d’énalapril/lercanidipine ont nécessité une hospitalisation. Les symptômes rapportés (diminution de la pression artérielle systolique, bradycardie, agitation, somnolence, douleur latérale) pourraient être également dus à l’administration concomitante de doses élevées d’autres médicaments (ex : β-bloquants).
Les caractéristiques les plus importantes du surdosage rapportées à ce jour avec l’énalapril sont une hypotension sévère (débutant environ 6 heures après l’ingestion des comprimés) associée à un blocage du système rénine-angiotensine et une stupeur.
D’autres symptômes associés à un surdosage d’IEC peuvent comporter : choc circulatoire, troubles électrolytiques, insuffisance rénale, hyperventilation, tachycardie, palpitations, bradycardie, sensations vertigineuses, anxiété et toux. Des concentrations sériques d’énalaprilate 100 et 200 fois supérieures à celles généralement observées après l’administration de doses thérapeutiques, ont été rapportées après l’ingestion de respectivement 300 mg et 440 mg d’énalapril.
Comme avec d’autres dihydropyridines, un surdosage en lercanidipine provoque une vasodilatation périphérique excessive avec hypotension marquée et tachycardie réflexe. Cependant, à très fortes doses, la sélectivité vasculaire n’est pas maintenue, provoquant une bradycardie et un effet inotrope négatif. Les effets indésirables les plus fréquents lors d’un surdosage sont l’hypotension, les sensations vertigineuses, les céphalées et les palpitations.
Le traitement recommandé du surdosage en énalapril est une perfusion intraveineuse de solution saline isotonique.
En cas d’hypotension, le patient doit être allongé les pieds surélevés. Si disponible, un traitement avec une perfusion d’angiotensine II et/ou des catécholamines par voie intraveineuse peut également être envisagé. Si l’ingestion des comprimés est récente, des mesures visant à éliminer le maléate d’énalapril doivent être prises (ex : vomissements, lavage gastrique, administration d’absorbants ou de sulfate de sodium). L’énalaprilate peut être éliminé de la circulation sanguine par hémodialyse (voir rubrique 4.4). L’utilisation d’un stimulateur cardiaque est indiquée en cas de bradycardie résistante au traitement. Les signes vitaux, les électrolytes sériques et les taux de créatinine doivent être surveillés en continu.
Pour la lercanidipine, une hypotension cliniquement significative nécessite une prise en charge cardio-vasculaire active avec une surveillance fréquente de la fonction cardiaque et respiratoire, une surélévation des jambes et un contrôle du volume liquidien circulatoire et du débit urinaire.
Compte tenu de l’action pharmacologique prolongée de la lercanidipine, l’état cardiovasculaire des patients en surdosage doit être surveillé pendant au moins 24 heures. Ce médicament étant fortement lié aux protéines plasmatiques, une dialyse sera vraisemblablement inefficace.
Classe pharmacothérapeutique : Inhibiteur de l’enzyme de conversion de l’angiotensine et inhibiteur calcique : énalapril et lercanidipine
Code ATC : C09BB02
ZANEXTRA® est une association fixe d’un inhibiteur de l’enzyme de conversion (énalapril) et d’un inhibiteur calcique (lercanidipine), deux composés antihypertenseurs avec un mécanisme d’action complémentaire permettant le contrôle de la pression artérielle chez les patients atteints d’une hypertension essentielle.
Le maléate d’énalapril est un sel de l’énalapril, un dérivé de deux acides aminés, la L-alanine et la L-proline. L’enzyme de conversion de l’angiotensine (ECA) est une peptidyl-dipeptidase qui catalyse la conversion de l’angiotensine I en angiotensine II, substance vasopressive.
Après absorption, l’énalapril est hydrolysé en énalaprilate qui inhibe l’ECA. L’inhibition de l’ECA entraîne une diminution de l’angiotensine II plasmatique, ce qui entraîne une augmentation de l’activité de la rénine plasmatique (par suppression du rétrocontrôle négatif de la sécrétion de rénine) et une diminution de la sécrétion d’aldostérone.
Puisque l’ECA est identique à la kininase II, l’énalapril peut également inhiber la dégradation de la bradykinine, un puissant peptide vasodépresseur. Cependant, le rôle de ce mécanisme dans les effets thérapeutiques de l’énalapril n’est pas encore compris.
Bien que le mécanisme par lequel l’énalapril réduit la pression artérielle soit principalement attribué à l’inhibition du système rénine-angiotensine-aldostérone, l’énalapril a un effet antihypertenseur même chez les patients ayant des concentrations de rénine basses.
L’administration d’énalapril à des patients hypertendus entraîne une diminution de leur pression artérielle en position débout et allongée, sans augmentation significative de leur fréquence cardiaque.
Une hypotension orthostatique symptomatique est rare. Chez certains patients, un contrôle optimal de la pression artérielle n’est obtenu qu’après plusieurs semaines de traitement. Une interruption brutale du traitement par énalapril n’est pas associée à une augmentation rapide de la pression artérielle.
Une inhibition efficace de l’activité de l’ECA se produit normalement entre 2 et 4 heures après l’administration orale d’une dose unique d’énalapril. Le début de l’effet antihypertenseur est généralement observé après une heure et la réduction maximale de la pression artérielle est observée entre 4 et 6 heures après l’administration. La durée de l’effet est dose-dépendante, cependant, aux doses recommandées, les effets antihypertenseurs et hémodynamiques persistent pendant au moins 24 heures.
Des études hémodynamiques chez des patients atteints d’hypertension artérielle essentielle ont démontré que la réduction de la pression artérielle était associée à une diminution de la résistance artérielle périphérique et une augmentation du débit cardiaque avec peu ou pas de modification de la fréquence cardiaque. Suite à l’administration d’énalapril, le débit sanguin rénal a augmenté tandis que le taux de filtration glomérulaire est resté inchangé. Il n’y a pas eu de signe de rétention sodée ou hydrique. Cependant, chez les patients ayant des taux faibles de filtration glomérulaire avant traitement, ces taux ont généralement augmenté.
Des études cliniques à court terme chez des patients diabétiques et non diabétiques ayant une affection rénale, ont mis en évidence des diminutions de l’albuminurie et de l’excrétion urinaire des IgG et de la protéinurie totale après l’administration d’énalapril.
Deux grands essais cliniques randomisés, contrôlés ONTARGET (Ongoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial) et VA NEPHRON-D (The Veterans Affairs Nephropathy in Diabetes) ont étudié l'utilisation d'une association d'un inhibiteur de l'enzyme de conversion et d'un antagoniste des récepteurs de l'angiotensine II.
L’étude ONTARGET a été réalisée chez les patients ayant des antécédents de maladie cardiovasculaire ou de maladie vasculaire cérébrale ou atteints d’un diabète de type 2 avec une atteinte des organes cibles.
L’étude VA NEPHRON-D a été réalisée chez des patients diabétiques de type 2 et atteints d'une néphropathie diabétique.
En comparaison à une monothérapie, ces études n’ont pas mis en évidence d’effet bénéfique significatif sur l’évolution des atteintes rénales et/ou cardiovasculaires et sur la mortalité, alors qu’il a été observé une augmentation du risque d’hyperkaliémie, d’insuffisance rénale aiguë et/ou d’hypotension.
Ces résultats sont également applicables aux autres IEC et ARA II, compte tenu de la similarité de leurs propriétés pharmacodynamiques.
Les IEC et ARA II ne doivent donc pas être associés chez les patients atteints de néphropathie diabétique.
L’étude ALTITUDE (Aliskiren Trial in Type 2 Diabetes Using Cardiovascular and Renal Disease Endpoints) a été réalisée dans le but d’évaluer le bénéfice de l’ajout d’aliskiren à un traitement standard par IEC ou un ARA II chez les patients atteints d’un diabète de type 2 et d’une insuffisance rénale chronique, avec ou sans troubles cardiovasculaires. Cette étude a été arrêtée prématurément en raison d’une augmentation du risque d’évènements indésirables. Les décès d’origine cardiovasculaire et les accidents vasculaires cérébraux ont été plus fréquents dans le groupe aliskiren que dans le groupe placebo ; de même les évènements indésirables et certains évènements indésirables graves tels que l’hyperkaliémie, l’hypotension et l’insuffisance rénale ont été rapportés plus fréquemment dans le groupe aliskiren que dans le groupe placebo.
La lercanidipine est un antagoniste calcique du groupe des dihydropyridines qui inhibe le flux transmembranaire de calcium vers le muscle cardiaque et les muscles lisses. Le mécanisme de l’effet antihypertenseur est basé sur un effet relaxant direct sur les muscles lisses vasculaires, diminuant ainsi la résistance périphérique totale. Malgré une demi-vie plasmatique courte, et grâce à son coefficient de partage membranaire élevé, la lercanidipine possède un effet antihypertenseur prolongé et est dépourvue d’effet inotrope négatif en raison de sa forte sélectivité vasculaire.
Puisque la vasodilatation est induite très progressivement par la lercanidipine, une hypotension aiguë avec tachycardie réflexe n’a été observée que dans de rares cas chez les patients hypertendus.
Comme avec d’autres 1,4-dihydropyridines asymétriques, l’effet antihypertenseur de la lercanidipine résulte principalement de son énantiomère S.
L'association de ces 2 molécules a un effet additif antihypertenseur, baissant la pression artérielle en façon plus importante que chacun des 2 composants pris séparément.
Dans une étude pivotale de phase III, en double aveugle, en add-on d’association thérapeutique, réalisée chez 342 patients non répondeurs à la lercanidipine 10 mg (définis par une pression artérielle diastolique comprise entre 95 et 114 mmHg et une pression artérielle systolique comprise entre 140 et 189 mmHg), la réduction de la pression artérielle systolique a été supérieure de 5,4 mmHg avec l’association énalapril 10 mg/lercanidipine 10 mg, par rapport au groupe sous lercanidipine 10 mg seule (- 7,7 mmHg versus - 2,3 mmHg, p < 0,001), après 12 semaines de traitement ; la réduction de la pression artérielle diastolique a été supérieure de 2,8 mmHg pour le groupe sous association fixe (- 7,1 mmHg versus - 4,3 mmHg, p < 0,001) pour le groupe sous monothérapie.
Le taux de répondeurs a été significativement supérieur avec l’association fixe par rapport à la monothérapie : 41% contre 24% (p < 0,001) pour la pression artérielle systolique et 35% contre 24%, (p = 0,032) pour la pression artérielle diastolique. Un pourcentage supérieur significatif de patients sous association fixe a présenté une normalisation de la pression artérielle systolique (39% versus 22%, p < 0,001) et de la pression artérielle diastolique (29% versus 19%, p = 0,023) par rapport aux patients sous monothérapie.
Pendant la phase de suivi à long terme de cette étude, réalisée en ouvert, une titration avec l’association fixe énalapril 20 mg/lercanidipine 10 mg était possible si la pression artérielle restait supérieure à 140/90 mmHg ; 133 patients sur 221 ont eu cette titration et la pression artérielle diastolique a été normalisée pour un tiers des cas.
Dans une étude pivotale de phase III, en double aveugle, de traitement complémentaire, réalisée chez 327 patients non répondeurs à l’énalapril 20 mg (définis par une pression artérielle diastolique comprise entre 95 et 114 mmHg et une pression artérielle systolique comprise entre 140 et 189 mmHg), la réduction de la pression artérielle systolique a été de -9,8 mmHg avec l’association énalapril 20 mg/lercanidipine 10 mg, par rapport au groupe sous monothérapie (- 6,7 mmHg, p = 0,013) ; la réduction de la pression artérielle diastolique a été de -9,2 mmHg pour le groupe sous association fixe contre -7,5 mmHg (p = 0,015) pour le groupe sous monothérapie.
Le taux de répondeurs n’a pas été significativement supérieur avec l’association fixe par rapport à la monothérapie : 53% contre 43% (p = 0,076) pour la pression artérielle diastolique et 41% contre 33% (p = 0,116) pour la pression artérielle systolique. Un pourcentage supérieur, mais non significatif de patients sous association fixe a présenté une normalisation de la pression artérielle diastolique (48% versus 37% (p = 0,055) et de la pression artérielle systolique (33% versus 28%, p = 0,325) par rapport aux patients sous monothérapie.
Dans une étude contrôlée, randomisée, en double aveugle, versus placebo et comparateur actif, avec un plan factoriel, menée sur 1 039 patients atteints d’hypertension artérielle modérée (définie par une pression artérielle diastolique (PAD) entre 100 et 109 mmHg et confirmée par une PAD ≥ 85 mmHg au domicile, et une pression artérielle systolique (PAS) < 180 mmHg en consultation), les patients sous énalapril 20 mg/lercanidipine 20 mg ont eu une diminution significative des pressions artérielles diastolique et systolique comparée au placebo (p < 0,001) en consultation et à domicile.
En consultation, des différences ont été observées dans la variation de la PAD par rapport à l’inclusion avec l’association 20 mg/20 mg (-15,2 mmHg, n = 113) comparativement à l’énalapril 20 mg seul (-11,3 mmHg, p = 0,004, n = 113) ou à la lercanidipine 20 mg seule (-13,0 mmHg, p = 0,092, n = 113).
De même, en consultation, des différences ont été observées dans la variation de la PAS par rapport à l’inclusion entre l’association 20 mg/ 20 mg (-19,2 mmHg) et la lercanidipine 20 mg seule (-13,0 mmHg, p = 0,002) ou l’énalapril 20 mg seul (-15,3 mmHg, p = 0,055). Des différences ont aussi été observées pour les pressions artérielles diastoliques et systoliques à domicile.
Le taux de patients répondeurs a été significativement supérieur avec l’association fixe par rapport au placebo (p < 0,001) et aux 2 monothérapies (p < 0,01) pour la PAD (75%) et la PAS (71%).
La normalisation de la pression artérielle a été atteinte par un pourcentage plus élevé de patients traités par l’association 20 mg/20 mg (42%) que par le placebo (22%).
Aucune interaction pharmacocinétique n’a été observée lors de l’administration concomitante d’énalapril et de lercanidipine.
L’énalapril, par voie orale, est rapidement absorbé, avec des pics de concentration plasmatique atteints en une heure. En se basant sur l’élimination urinaire, la quantité d’énalapril absorbée après administration orale de comprimés est d’environ 60%. L’absorption d’énalapril par voie orale n’est pas influencée par la présence d’aliments dans le tube digestif.
Après absorption par voie orale, l’énalapril est rapidement et largement hydrolysé en énalaprilate, un puissant inhibiteur de l’enzyme de conversion de l’angiotensine. Des pics de concentrations sériques d’énalaprilate sont atteints environ 4 heures après la prise d’un comprimé d’énalapril par voie orale. La demi-vie efficace d’accumulation de l’énalaprilate après administration de doses multiples d’énalapril par voie orale est de 11 heures. Chez les patients avec une fonction rénale normale, la concentration plasmatique à l'état d'équilibre est atteinte après 4 jours de traitement.
Dans la fourchette de concentrations liées aux doses thérapeutiques, la liaison aux protéines plasmatiques de l’énalaprilate chez l’homme ne dépasse pas 60%.
Hormis la transformation en énalaprilate, il n’a pas été mis en évidence de métabolisme significatif de l’énalapril.
L’élimination de l’énalaprilate est essentiellement rénale. Les principaux composés retrouvés dans l’urine sont l’énalaprilate, qui représente environ 40% de la dose, et de l’énalapril sous forme inchangée (environ 20%).
L’exposition à l’énalapril et l’énalaprilate est augmentée chez les patients atteints d’insuffisance rénale. Après administration de 5 mg/jour chez des patients ayant une insuffisance rénale légère à modérée (clairance de la créatinine entre 40 et 60 ml/min), l’AUC de l’énalaprilate à l’état d’équilibre a été environ deux fois plus élevée que chez les patients ayant une fonction rénale normale. En cas d’insuffisance rénale sévère (clairance de la créatinine ≤ 30 ml/min), l’AUC a été augmentée d’environ 8 fois. La demi-vie effective de l’énalaprilate après administration de doses multiples d’énalapril est prolongée à ce degré d’insuffisance rénale et le délai pour atteindre l’état d’équilibre est retardé (voir rubrique 4.2). L’énalaprilate peut être éliminé de la circulation sanguine par hémodialyse, à une clairance de 62 ml/min.
Après administration d’une dose unique de 20 mg d’énalapril administré par voie orale chez 5 femmes allaitant, les concentrations maximales moyennes d’énalapril dans le lait étaient de 1,7 µg/L (0,54 µg/L à 5,9 µg/L), 4 à 6 heures après la prise. Les concentrations maximales moyennes d’énalaprilate dans le lait étaient de 1,7 µg/L (1,2 µg/L à 2,3 µg/L) ; ces concentrations maximales étaient obtenues à des moments divers au cours de la période de 24 h.
A partir de ces données observées dans le lait maternel, on estime qu’un enfant allaité exclusivement à partir du lait maternel serait exposé à une dose maximale correspondant à 0,16% de la dose quotidienne de la mère après ajustement au poids. Une femme qui avait reçu par voie orale 10 mg par jour d’énalapril pendant 11 mois présentait des concentrations maximales d’énalapril dans le lait de 2 µg/L, 4 heures après la prise et des concentrations maximales d’énalaprilate dans le lait de 0,75 µg/L, 9 heures environ après la prise.
La concentration totale d’énalapril et d’énalaprilate mesurée dans le lait au cours de la période de 24 heures était de respectivement 1,44 µg/L et de 0,63 µg/L, la concentration d’énalaprilate n’était plus détectable dans le lait (< 0,2 µg/L) 4 heures après l’administration d’une dose unique de 5 mg d’énalapril chez une mère et de 10 mg d’énalapril chez 2 mères. Les concentrations d’énalapril n’ont pas été dosées.
La lercanidipine est totalement absorbée après administration orale et les pics de concentration plasmatique sont atteints après 1,5 à 3 heures environ.
Les deux énantiomères de la lercanidipine ont un profil pharmacocinétique similaire. Le délai pour atteindre le pic plasmatique est identique et le pic plasmatique et l’AUC sont, en moyenne, 1,2 fois plus élevés pour l’énantiomère S. La demi-vie d’élimination des deux énantiomères est pratiquement identique. Aucune interconversion des deux énantiomères n’a été observée in vivo.
En raison de l’effet de premier passage important, la biodisponibilité absolue de lercanidipine après administration orale et après prise de nourriture est d’environ 10%. Cependant, la biodisponibilité après administration à des volontaires sains à jeun est réduite à 1/3.
La biodisponibilité orale de la lercanidipine augmente de 4 fois lorsqu’elle est administrée dans les 2 heures suivant un repas riche en graisses. Par conséquent, le médicament doit être pris avant les repas.
La distribution depuis le plasma dans les tissus et organes est rapide et importante.
La liaison de la lercanidipine aux protéines plasmatiques est supérieure à 98%. Comme les taux de protéines plasmatiques sont diminués chez les patients atteints d’une insuffisance rénale ou hépatique sévère, la fraction libre du médicament peut être plus élevée chez ces patients.
La lercanidipine est largement métabolisée par le CYP3A4. On ne retrouve aucune substance mère dans l’urine ou les selles. La lercanidipine est principalement métabolisée en métabolites inactifs et environ 50% de la dose est éliminée dans l’urine.
Les essais in vitro sur des microsomes hépatiques humains ont démontré que la lercanidipine montre une légère inhibition des deux enzymes CYP3A4 et CYP2D6 à des concentrations 160 fois et 40 fois supérieures aux pics de concentration plasmatique obtenus après l’administration d’une dose de 20 mg.
De plus, des études d’interaction chez l’homme ont montré que la lercanidipine ne modifie pas les concentrations plasmatiques du midazolam, un substrat typique du CYP3A4 ou du métoprolol, un substrat typique du CYP2D6. Par conséquent, aux doses thérapeutiques, une inhibition par la lercanidipine de la biotransformation des substances métabolisées par le CYP3A4 ou le CYP2D6, n’est pas attendue.
L’élimination se fait essentiellement par biotransformation.
La demi-vie d’élimination terminale moyenne est de 8 à 10 heures, et du fait de la forte liaison aux membranes lipidiques, l’effet thérapeutique dure 24 heures. Aucune accumulation n’a été mise en évidence après administration répétée.
L’administration orale de la lercanidipine entraîne des concentrations plasmatiques non directement proportionnelles à la dose (cinétique non linéaire). Après l’administration de 10, 20 ou 40 mg, les pics de concentration plasmatique étaient dans les proportions de 1:3:8 et les aires sous les courbes des concentrations plasmatiques en fonction du temps dans les proportions de 1:4:18, ce qui évoque une saturation progressive de l’effet de premier passage. Par conséquent, la biodisponibilité augmente avec l’accroissement de la dose.
Le comportement pharmacocinétique de la lercanidipine chez les patients âgés et chez les patients atteints d’insuffisance rénale ou hépatique légère à modérée est similaire à celui observé dans la population générale. Des concentrations plus élevées du médicament (environ 70%) ont été mises en évidence chez les patients atteints d’insuffisance rénale sévère ou dialysés. Chez les patients atteints d’insuffisance hépatique modérée à sévère, la biodisponibilité systémique de la lercanidipine est probablement augmentée car le médicament est largement métabolisé par le foie.
La toxicité potentielle de l’association fixe d’énalapril et de lercanidipine a été étudiée chez le rat après administration orale et ce jusqu’à 3 mois, lors de 2 études génotoxiques. L’association n’a pas modifié le profil toxicologique de chacun des 2 composants.
Les données suivantes sont disponibles pour chaque composant (énalapril et lercanidipine) :
Les données précliniques ne révèlent aucun risque particulier pour l’homme d’après les études conventionnelles de pharmacologie sur la sécurité d’emploi, de toxicité à doses répétées, de génotoxicité et de potentiel carcinogène.
Les études de toxicité sur la reproduction suggèrent que l’énalapril n’a aucun effet sur la fertilité et les performances reproductives chez le rat, et qu’il n’est pas tératogène. Dans une étude au cours de laquelle des rates ont été traitées avant la période allant de l’accouplement à la gestation, une augmentation de l’incidence de la mortalité de la descendance est survenue au cours de l’allaitement. Il a été montré que ce composé traversait la barrière placentaire et était excrété dans le lait maternel. Les IEC induisent globalement des effets indésirables sur le développement fœtal tardif, provoquant une mortalité fœtale et des effets congénitaux, en particulier au niveau du crâne. Une fœtotoxicité, des retards de croissance intra-utérins et une persistance du canal artériel ont été notifiés.
Ces anomalies du développement semblent être dues pour partie à l’action directe des IEC sur le système rénine-angiotensine du fœtus, et pour partie à l’ischémie provenant de l’hypotension maternelle et à la diminution de l’irrigation sanguine fœto-placentaire avec pour conséquence une réduction de l’apport en oxygène et en nutriments au fœtus.
Les données précliniques ne révèlent aucun risque particulier pour l’homme d’après les études conventionnelles de pharmacologie sur la sécurité d’emploi, de toxicité à doses répétées, de génotoxicité et de potentiel carcinogène.
Des effets significatifs observés au cours d’études à long terme chez le rat et le chien ont été imputés, directement ou indirectement, aux effets connus de doses élevées d’inhibiteurs calciques, reflétant principalement une activité pharmacodynamique exagérée.
Le traitement par la lercanidipine n’a eu aucun effet sur la fertilité ou les performances générales de reproduction chez le rat, mais à hautes doses, il induit des pertes survenant avant et après l’implantation et des retards du développement fœtal. Il n’existe aucun élément attestant d’un effet tératogène chez le rat et le lapin, mais d’autres dihydropyridines ont provoqué des effets tératogènes chez l’animal. La lercanidipine a induit une dystocie lorsqu’elle a été administrée à doses élevées (12 mg/kg/jour) au cours de la parturition.
La distribution de la lercanidipine et/ou de ses métaboliques chez les femelles gestantes et leur excrétion dans le lait maternel n’a pas été étudiée chez l’animal.
Noyau : Lactose monohydraté, cellulose microcristalline, carboxyméthylamidon sodique type A, povidone K30, bicarbonate de sodium, stéarate de magnésium.
Pelliculage : Hypromellose 5cP, dioxyde de titane (E171), talc, macrogol 6000, laque aluminique de jaune de quinoléine (E104), oxyde de fer jaune (E172).
Sans objet.
2 ans.
A conserver dans l’emballage extérieur d’origine pour protéger le médicament de la lumière et de l’humidité.
A conserver à une température ne dépassant pas 25°C.
Plaquettes (Polyamide-Aluminium-PVC/Aluminium).
Boîte de 7, 14, 28, 30, 35, 42, 50, 56, 90, 98 et 100 comprimés.
Toutes les présentations peuvent ne pas être commercialisées.
Tout médicament non-utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
BOUCHARA-RECORDATI
Immeuble "Le Wilson"
70, avenue du Général de Gaulle
92800 Puteaux
FRANCE
Liste I.
N/A
N/A
Sans objet.
Sans objet.
Dernière mise à jour de cette page
14/10/2020.
![]() Adresse
Adresse
![]() Téléphone
Téléphone
Revue MAN
Revue OST
Actualités
Webinaires
Espaces labos
Site éditeur :
![]()